In many experimental designs we want to map the genetic loci responsible for viability selection. Often this is done by sequencing "surviving" individuals in a genetic cross, e.g. F2 cross. Marker loci underlying the selected traits should show allele frequencies shifted from expectations.
Our idea was that you can increase the power of this analysis by not only sequencing those individuals that were selected for but also those that were selected against.
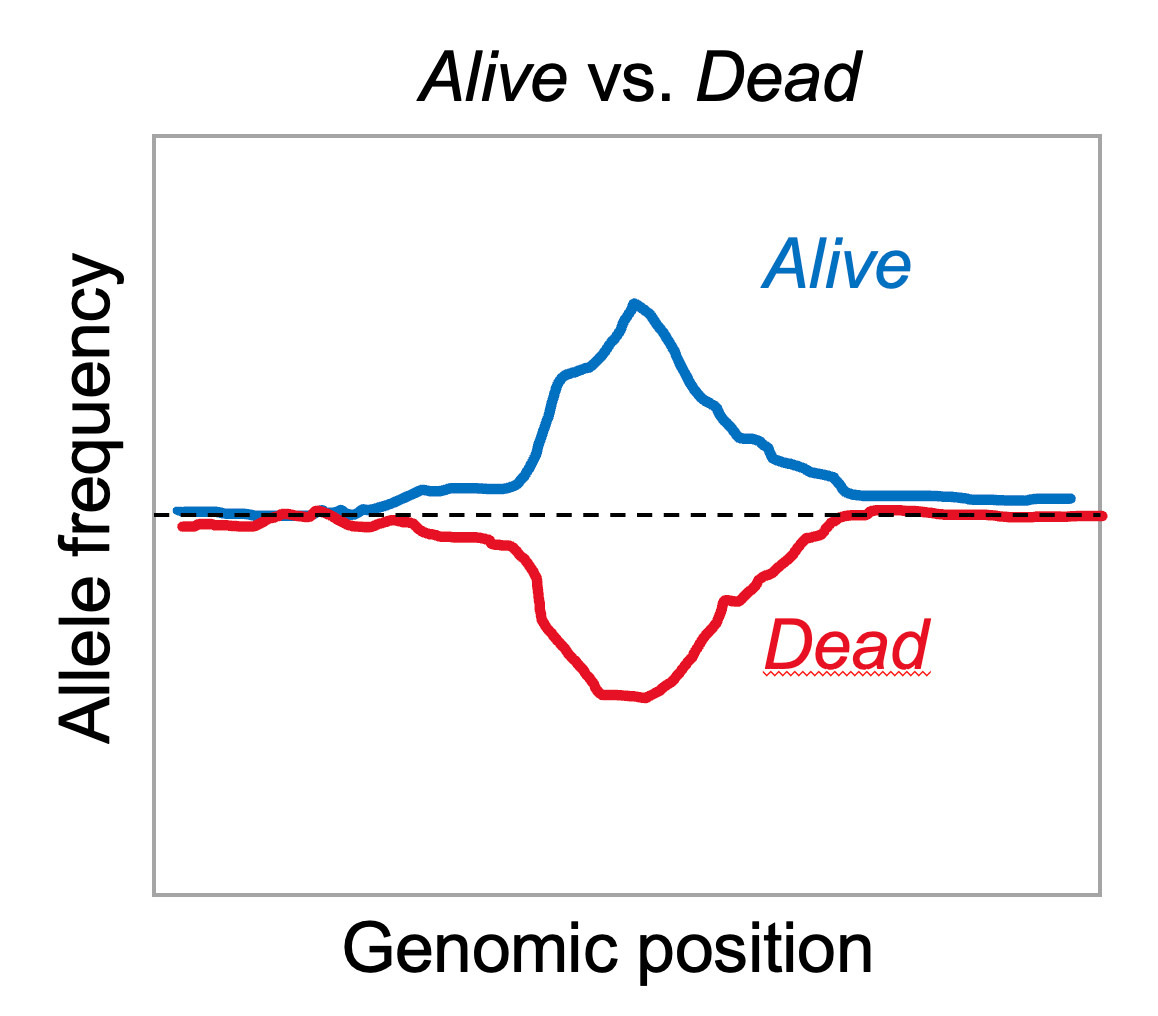
In the ideal case, this should create almost mirror images in allele frequency as exemplified in the plot below.
Ideally, you would individually sequence all individuals. However, this is still expensive and thus we sequenced pools of individuals. For anyone interested in trying a similar method, I would recommend sequencing several pools of Alive and Dead groups to reduce noise in the data.
If you are extra interested in the false positive rate under our implementation. Do check out our accompanying simulation study in the supplements (Text S1).
While we used the Alive vs Dead method to find putative loci involved in hybrid incompatibilities, it is applicable to any kind of viability selection.

Comments
In the ideal case, this should create almost mirror images in allele frequency as exemplified in the plot below.
https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1111%2Fmec.17672&file=mec17672-sup-0001-Supinfo.pdf
Thanks to reviewers for input on the method and Lima and Willet 2018 for inspiration.
https://academic.oup.com/jhered/article/109/4/469/4796873